
Les facteurs constitutionnels
L'âge
L'incidence du cancer colorectal augmente avec l'âge et devient appréciable à partir de 45 ans et double ensuite à chaque décennie.
Chez l'homme, l'incidence passe de 0,3 % pour les sujets de moins de 50 ans à 4,5 % pour les patients de 70 ans et plus. Chez la femme les taux vont, pour les mêmes tranches d'âge, de 0,1 % à 2,5 %.
Les formes familiales simples de cancer du côlon sont rares
Les formes dites sporadiques sont beaucoup plus fréquentes et représentent de 70 à 80 % des cas, selon les études. Les formes familiales sont beaucoup moins fréquentes et ne concernent que 10 à 30 % des cas. Ce sont des familles dans lesquelles on retrouve plusieurs cas de cancers du côlon sans notion d’hérédité nette. Dans cette situation, la maladie apparaît au même âge que pour les formes dites sporadiques et, à l'occasion d'une coloscopie, le gastro-entérologue trouve peu de polypes, voire aucun...
Les différentes formes familiales de cancers colorectaux sont :
- Une agrégation familiale est retrouvée dans environ 10 % des cas
- La polypose adénomateuse familial (PAF), de transmission héréditaire autosomique dominante qui contribue pour moins de 1 % des cas
- Les cancers colorectaux héréditaires sans polypose (HNPCC), de transmission héréditaire autosomique, qui représentent 10 à 15 % des cas
Des antécédents personnels de cancer
Une personne déjà traitée pour un cancer du côlon présente un risque plus élevé de développer un second cancer colorectal.
Les femmes ayant eu un cancer du sein, de l’ovaire ou de l’utérus ont également un risque un peu plus important de présenter un cancer colorectal.
Les principales formes héréditaires des cancers colorectaux
|
Syndrome |
Gène(s) en cause |
Mode de transmission |
|---|---|---|
|
Formes polyposiques |
||
|
Polyposes adénomateuses |
|
|
|
Polypose adénomateuse familiale |
APC |
Autosomique dominant |
|
Polypose associée à MUTYH |
MUTYH (MYH) |
Autosomique récessif |
|
Polyposes hamartomateuses |
|
|
|
Polypose juvénile |
SMAD4 ; BMPR1A |
Autosomique dominant |
|
Syndrome de Peutz-Jeghers |
STK11/LKB1 |
Autosomique dominant |
|
Formes non polyposiques |
||
|
Syndrome de Lynch (HNPCC) |
MLH1, MSH2, MSH6, PMS2 |
Autosomique dominant |
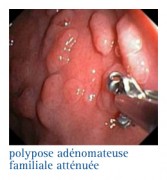
Les polyposes coliques
LA POLYPOSE ADÉNOMATEUSE FAMILIALE (PAF)
La polypose adénomateuse familiale est responsable d'environ 1 % des cancers colorectaux. C’est une maladie héréditaire de transmission autosomique dominante dont la pénétrance est très élevée, 90 % des cas.
Le syndrome de Gardner, associe à la polypose colique, la présence de tumeurs desmoïdes (tumeurs bénignes des tissus mous), des tumeurs bénignes cutanées et osseuses.
Elle est la conséquence d’une mutation constitutionnelle du gène suppresseur de tumeurs APC porté par le chromosome 5q. Cette mutation fut isolée en 1991.
Ainsi, chez un patient ayant une PAF, chaque cellule colique a déjà, de façon innée, inactivée une copie de ce gène suppresseur de tumeur. L'inactivation de la seconde copie est l'événement clef de la cancérogenèse colique, le développement de la maladie est donc beaucoup plus rapide.
Le risque de cancer est d’environ 50 % à 40 ans et pratiquement 100 % à l’âge de 60 ans. De fait, dans la PAF, le risque de cancer colorectal survient à un âge beaucoup plus jeune que pour les patients ayant une tumeur sporadique. Il existe des formes atténuées pour lesquels le risque est moins élevé.
Les polypes apparaissent généralement au cours de la deuxième décennie ; leur nombre croît avec l’âge.
Les polyposes colorectales sont de reconnaissance aisée dans la mesure où le diagnostic est établi sur la base des seules données de la coloscopie. Son aspect, en coloscopie, est illustré par l'image photographie de droite.
Elles sont toujours le reflet d’une prédisposition génétique et constituent une indication de consultation de génétique oncologique. De plus, une recherche génétique pour les membres de votre famille, si vous avez plus de 100 polypes à la coloscopie ou si vous avez une forme atténuée de PAF, est recommandée par certains spécialistes.
Du fait du grand nombre de polypes, leur exérèse endoscopique n'est pas techniquement possible.
La chirurgie prophylactique est généralement indiquée au cours de la troisième décennie. Il peut s’agir, en fonction de l’existence ou non d’une atteinte rectale et de sa sévérité, soit d’une colectomie subtotale avec anastomose iléo-rectale, soit d’une colo-proctectomie avec anastomose colo-anale.
IMPORTANT...
Des informations plus détaillées sur cette affection peuvent être trouvées sur le site Internet de l’Association pour la Prévention, le Traitement et l’Étude des Polyposes Familiales (APTEPF) 36, chemin de Grandchamp 39570 Courbouzon (Tél. 03 84 43 08 41 - aptepf@aol.com).
LES POLYPOSES HAMARTOMATEUSES
Un hamartome est une malformation due à une anomalie du développement avant la naissance.
Ce sont des affections très rares, dix fois moins fréquentes que la polypose adénomateuse familiale. Trois entités sont décrites par les spécialistes et sont, dans tous les cas, à transmission autosomique dominante :
- La polypose juvénile est liée à une mutation constitutionnelle des gènes SMAD4 ou BMPR1A
- Le syndrome de Peutz-Jeghers est la conséquence d'une mutation constitutionnelle du gène STK1 et se manifeste par une polypose digestive et des manifestations cutanéo-muqueuses. Il est aussi associé à une majoration du risque de cancer du sein, du pancréas, du poumon et d’adénocarcinome du col utérin.
- La maladie de Cowden est associée à une mutation constitutionnelle du gène PTEN
Dans ces cas, le risque cumulé au cours de l’existence de cancer colorectal est évalué à 40 à 50 % pour la polypose juvénile et le syndrome de Peutz-Jeghers. La polypose juvénile est également associée à une majoration du risque de cancer gastrique.
Les formes non polyposiques - syndrome de Lynch
DE QUOI S'AGIT-IL ?
Le syndrome de Lynch, sont parfois désignés par leurs initiales anglaises, HNPCC ( Hereditary Non Polyposis Colorectal Cancer ) est une forme héréditaire non polyposique de cancers colorectaux, pourvoyeuse de 3 % environ de l’ensemble des cancers colorectaux. .
C'est une maladie génétique de transmission autosomique dominante et représente la forme la plus fréquente de cancer colorectal héréditaire. Elle touche surtout le côlon droit.
Un petit retour en arrière...
La première description par Aldred Warthin (1866 - 1931 - pathologiste américain) du syndrome HNPCC date de 1913. Elle sera complétée en 1966 par Henri T. Lynch (médecin américain 1928 - 2019) qui a donné son nom au syndrome.
Cette prédisposition au cancer est définie cliniquement par les critères d'Amsterdam en 1991, modifiés en critères d'Amsterdam II en 1999, qui réunissent des informations individuelles et généalogiques :
- Au moins trois sujets atteints de cancers appartenant au spectre étroit du syndrome HNPCC (côlon, rectum, endomètre, intestin grêle, voies urinaires) et histologiquement prouvés
- Unis deux à deux par un lien de parenté au premier degré sur deux générations
- Un des cancers au moins s'étant révélé avant l'âge de 50 ans
Il existe deux variantes : le syndrome de Muirr Torre dont le spectre est élargi aux tumeurs cutanées sébacées et aux kérato-acanthomes et le syndrome de Turcot dont le spectre inclus le cancer du côlon et des tumeurs du système nerveux central (glioblastome, médulloblastome, épendymone)
LE RISQUE
Sa fréquence n'est pas parfaitement connue. Selon les études, en utilisant les critères d'Amsterdam, on estime qu'entre 3 et 5 % des cancers colorectaux seraient en rapport avec cette maladie.
Les familles touchées par ce groupe de maladies ont un risque plus élevé de développer certains types de cancers
LA TRANSMISSION
Il se fait sur le mode autosomal dominant. Les membres de ces familles ont un risque plus élevé, de 4 à 13 %, selon les études, de développer des cancers coliques.
L’âge d’apparition de la maladie se situe vers 45 ans, plus précoce que dans les formes non héréditaires.
LES GÈNES IMPLIQUÉS
Le système MMR (MisMach Repair) est un système physiologique de réparation des mésappariements de l’ADN lors de la réplication de l'ADN à l'occasion d'une division cellulaire.
Trois gènes principaux sont impliqués dans cette réparation : hMLH1, hMSH2 et hMSH6. Ce sont tous des gènes suppresseurs de tumeur.
En cas de mutations ou d'inactivation de ces gènes, la réparation de l’ADN n'est plus possible.Les micro-satellites sont des séquence d’ADN qui se présentent sous forme d'un motif nucléotidique (1 à 6 bases) répété. Le nombre de répétitions est variable selon les individus.
Lors de la réplication de l’ADN les erreurs sont fréquentes au niveau des microsatellites. Ces erreurs sont normalement réparées par les systèmes de réparation de l’ADN dont le système MMR.
La recherche d’un tel phénotype tumoral (phénotype MSI pour Microsatellite Instability , ou dMMR pour deficient MMR) est essentiel pour poser le diagnostic de la maladie.
Les formes héréditaires non-polyposiques sont de reconnaissance plus difficile puisque l'aspect des lésions lors de la coloscopie est celui des cancers colorectaux sporadiques. Néanmoins, elle est soupçonnée dans les cas suivants :
- Un âge inhabituellement jeune, inférieur à 60 ans, lors du diagnostic
- La présence de lésions multiples, synchrones ou métachrones
- Une agrégation familiale de cancers colorectaux ou de cancers faisant partie du spectre du syndrome (cancers de l’endomètre, des ovaires, de l’intestin grêle, des voies excrétrices urinaires, de l’estomac et des voies biliaires)
Une consultation d'oncogénétique est recommandée par les sociétés savantes, dans le cas suivants :
- Deux parents atteints d’un cancer du spectre dont un avant l’âge de 50 ans
- Un antécédent personnel de cancer du spectre HNPCC/Lynch
- Age < 40 ans
- Présence d’une instabilité microsatellitaire chez un patient de moins de 60 ans ou pour tout patient, quel que soit son âge, présentant un cancer du spectre HNPCC/Lynch et ayant un antécédent familial de cancer du spectre HNPCC/Lynch
Les conséquences de mutations constitutionnelles des gènes MMR
Le risque cumulé de développer un cancer colorectal avant l’âge de 70 ans est de 30 à 48 % selon les études. De plus, ces mutations peuvent jouer un rôle dans la genèse d’autres cancers touchant un grand nombre d’organes (cancers du spectre du syndrome de Lynch) : l'endomètre principalement (risque cumulé de 30-54 % avant 70 ans) et les voies biliaires, voies excrétrices urinaires (bassinet et uretère essentiellement), ovaires et intestin grêle ensuite estomac et tumeurs sébacées
@ Une association pour vous aider : HNPCC-Lynch association créée pour aider les patients souffrant de cette affection. 56, avenue Bosquet 75007 Paris - Courriel : contact@hnpcc-lynch.com
Pourquoi rechercher un cancer du colon HNPCC ?
5% des cancers du colon
Transmission du risque de cancer: autosomique dominant
Diagnostic important pour: la surveillance et le diagnostic pré-symptomatique des apparentés
Les autres maladies héréditaires
Le risque de cancer colorectal est aussi plus élevé dans de très rares maladies héréditaires.
- Le syndrome de Gardner qui serait une forme particulière de FAP
- Le syndrome de Turcot
- La maladie de Recklinghausen
@ Pour en savoir plus : LES FACTEURS GÉNÉTIQUES
Pour nous résumer, les facteurs de risque génétiques...
Une polypose adénomateuse familiale (PAF)
Un syndrome de Lynch (HNPCC)
Des cas dans la famille
Un premier cancer du côlon ou des polypes coliques
Un cancer génital, en particulier de l'endomètre ou un cancer du sein
Les niveaux de risque
DÉFINITION
Les spécialistes définissent plusieurs niveaux de risque, en fonction de critères précis. Cette classification permet de préciser le type de surveillance que l’on vous proposera. Les modalités de cette surveillance vous seront précisés dans le chapitre Dépistage.
CONCRÈTEMENT...
Les sujets à risque « faible » ou « moyen »
Il n'y a pas de cas de cancer colorectal dans votre famille. Dans ce cas vous êtes considérés, après l'âge de 50 ans, comme à risque « faible » ou « moyen ».
La population à « haut risque » comprend :
- Les patients dans les antécédents desquels on relève un cancer colorectal, un polype de plus de 1 cm de diamètre ou un polype comportant des éléments villeux ou, au moins deux adénomes rectocoliques.
- Les sujets ayant un antécédent familial au 1er degré, père, mère, frères, sœurs, de cancer colorectal.
- Les patients atteints d'une rectocolite hémorragique ou d'une maladie de Crohn colique étendues et anciennes.
La population à « très haut risque »
Sont considérés comme à très haut risque, les personnes appartenant aux familles où existent, soit une polypose rectocolique familiale (FAP), soit un syndrome de Lynch (HNPCC) définit comme suit :
- Au moins 3 parents atteints d'un cancer colorectal ou d'un cancer épidémiologiquement lié, comme un cancer du sein, de l’utérus, du pancréas, dont un est parent au premier degré des deux autres,
- Au moins deux générations touchées,
- Un cancer découvert avant l'âge de 50 ans.
Schématiquement..
| JE SUIS SEUL DANS MA FAMILLE A ÊTRE ATTEINT... Quel risque pour les membres de la famille ? |
DES MEMBRES DANS MA FAMILLE ONT EU UN CANCER COLORECTAL... |
|---|---|
|
Augmentation du risque |
Peut-être une forme héréditaire, si votre père ou votre mère a eu
Attitude pratique : surveillance à partir de 40 ans, avec coloscopies régulières |
Quand faut-il envisager une consultation d'oncogénétique ?
≥ 15 polypes synchrones ou métachrones (comptés sur les coloscopies successives) quel que soit l’âge
≥ 10 polypes synchrones ou métachrones avant 60 ans
≥ 5 polypes synchrones ou métachrones avec au moins un des critères suivants
-
Avant 40 ans
-
Associés à un cancer colorectal MSS avant 60 ans
-
Tous avancés (> 1 cm, composante villeuse, dysplasie de haut grade
-
Adénomes duodénaux
-
Atteinte cutanée sébacée (adénome ou carcinome sébacé)
-
Hyperplasies sébacées multiples ou de grande taille avant 50 ans
Polypose (>100 polypes)
-
Atteinte sur plusieurs générations ou de tumeur desmoïde (recherche d’une mutation du gène APC)
-
En cas de phénotype atténué en l’absence d’agrégation transgénérationnelle, l’analyse débutera par la recherche d’une mutation du gène MUTYH
Mise à jour
31 mars 2021