
Le rétinoblastome
UNE TUMEUR RARE DE L'ŒIL…
ÉPIDÉMIOLOGIE
Le rétinoblastome est une tumeur maligne, rare, de la rétine mais la plus fréquente chez l’enfant.
Son incidence est de 1/15 000 à 1/20 000 naissances, avec une légère prédominance chez les garçons.
Elle apparaît habituellement avant l'âge de 5 ans. On estime à 8000 cas à travers le monde.
SES CARACTÉRISTIQUES
Dans environ 60 % des cas, la tumeur est unilatérale avec un âge médian au diagnostic de 2 ans et la plupart de ces formes sont non héréditaires.
Dans 40 % des cas, la tumeur est bilatérale et est toujours héréditaire, mais pas forcément familiales. Il est alors découvert à un âge médian plus précoce vers 1 an.
LES FORMES FAMILIALES
Le rétinoblastome est familial dans 10 % des cas, et dans ce cas le plus souvent bilatéral et multifocal, de révélation précoce (la moitié avant l'âge d'un an) avec une pénétrance élevée, de plus de 90 %.
La maladie se comporte dans ce cas comme une maladie autosomique dominante.
Les origines de la maladie
LE BLASTÈME
C'est un amas de cellules mésoblastiques (cellules du troisième feuillet embryonnaire) non différenciées donnant naissance à un organe ou à une partie du corps. Par exemple, le blastème dit métanéphrotique donnera naissance au rein, le blastème de membre sera à l'origine du bras ou de la jambe.
UN RÉTINOBLASTOME
C'est une tumeur de la rétine qui se développe à partir de cellules immatures dérivées de l’ébauche embryonnaire, le blastème, d'où le suffixe blastome . Elle correspond à la prolifération de petites cellules rondes dans la rétine.
LES ANOMALIES GÉNÉTIQUES
Le gène RB1 est situé sur le chromosome 13 (13q14). C'est un gène suppresseur de tumeur. Sa délétion aboutit au développement du rétinoblastome.
Dans les familles où ce cancer est fréquent, une portion du chromosome manque sur l'un des deux chromosomes dans toutes les cellules, en particulier celles de la rétine de l'œil. Ces cellules, à ce stade sont encore normales. Elles ne deviennent cancéreuses que lorsque la même région est perdue sur l'autre chromosome 13.
Dans les formes non héréditaires, les mutations des deux allèles surviennent spontanément au niveau d’une seule cellule de la rétine.
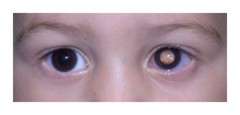
LE DIAGNOSTIC
LES SIGNES & SYMPTÔMES...
Il y a deux signes qui évoquent le diagnostic :
- Une leucocorie ou œil de chat est-à-dire, un reflet de la pupille blanchâtre, que l'on peut aisément repérer sur une photo
- Un strabisme
Ces symptômes, mêmes intermittents, nécessitent une consultation urgente d’ophtalmologie avec un examen du fond d’œil.
D’autres signes sont plus rares : buphtalmie (augmentation de la taille de l'œil), rubéose irienne (apparition de néo-vaisseaux), hypopion (accumulation de pus dans la chambre antérieure de l’œil), hyphéma (présence de sang dans la chambre antérieure de l'œil) et/ou une baisse d’acuité visuelle.
LE DIAGNOSTIC
Le fond d’œil
Il reste essentiellement clinique, fondé sur l’examen du fond d’œil sous anesthésie générale en milieu spécialisé avec visualisation de la lésion tumorale blanche, hypervascularisée. Cet examen permet le bilan précis des lésions intra-oculaire.
L'imagerie médicale
Une imagerie orbitaire et cérébrale est toujours nécessaire au diagnostic pour le bilan d’extension et la recherche d’anomalies intracrâniennes éventuellement associées. L’imagerie par résonance magnétique (IRM) est actuellement l’examen de référence et doit être réalisée avec sédation ou idéalement sous anesthésie générale pour en garantir la qualité.
La stadification et ses conséquences
LES VALEURS pTNM
La formule la plus précise en termes de pronostic et de traitements complémentaires après la thérapie curative est la classification anatomo-pathologique (p).
La tumeur
- pT1 : la tumeur confinée à la rétine, l' espace sous-rétinien et le vitré. Il n'y a pas d’atteinte du nerf optique, ni de la choroïde. Le traitement curatif ne sera suivi d'un traitement adjuvant.
- pT2 : il y a un envahissement minime du nerf optique et/ou de ses gaines méningées :
- pT2a correspond à une atteinte de la lame criblée (partie de l'os ethmoïde situé à la base du crâne) mais sans la dépasser
- pT2b traduit un envahissement focal de la choroïde
- pT2c désigne les cas présentant à la fois les caractéristiques a et b Le traitement curatif ne sera suivi d'un traitement adjuvant. Le pronostic vital excellent.
- pT3 : il existe un envahissement significatif du nerf optique et/ou de ses gaines méningées
- pT3a correspond à un envahissement retrolaminaire avec tranche de section saine En plus du traitement curatif, une chimiothérapie est recommandée. Le pronostic est plus réservé.
- pT3b décrit un envahissement massif de la choroïde En plus du traitement curatif, une chimiothérapie est recommandée ou la participation à un essai thérapeutique
- pT3c désigne les cas correspondant à a+b
- pT4 Il existe une extension de la maladie en dehors de l'œil. Le traitement chirurgical est complété par une chimiothérapie associée ou non à une radiothérapie, curiethérapie ou externe.
Les ganglions (N)
- pN0 correspond à l'absence de ganglions touchés.
- pN1 indique la présence d'adénopathies satellites de la tumeur au niveau de la tête ou du cou.
Les métastases (M)
- pM0 correspond à l'absence de métastases à distance.
- pM1 indique la présence de métastases soit dans le système nerveux central ou à distance
- pM1a dans la moelle osseuse
- pM1b dans d'autres sites
LES QUATRE STADES DE LA MALADIE
| Stade | Caractéristiques |
|---|---|
| 0 | Œil accessible à un traitement conservateur |
| I | Œil énucléé avec résection microscopiquement complète |
| II | Œil énucléé avec résection microscopiquement incomplète |
| III |
Extension régionale |
| IV |
Extension métastatique |
LES TRAITEMENTS
EN CENTRE SPECIALISÉ UNIQUEMENT !
Ils sont réalisés en centres spécialisés et comprennent diverses modalités, non exclusives et souvent complémentaires, comme :
- La chirurgie avec parfois une ablation chirurgicale de l'œil (énucléation)
- La chimiothérapie (carboplatine/étoposide avec deux cycles espacés de 21 jours)
- Le laser diode, la thermo-chimiothérapie, la cryothérapie, la photocoagulation.
- La radiothérapie
LES GRANDES OPTIONS
En cas de rétinoblastome unilatéral étendu
Dans ce cas, la plupart des patients doivent aujourd’hui encore être traités par une énucléation, car l’extension intra-oculaire de la tumeur ne permet pas d’envisager un traitement conservateur.
En l’absence d’envahissement du nerf optique ou des enveloppes oculaires qui est le cas dans la majorité des patients, aucun traitement adjuvant n’est indiqué.
Lorsque l’exérèse est microscopiquement incomplète, une chimiothérapie postopératoire et une irradiation orbitaire doivent être réalisées.
Les traitements conservateurs
C'est l'option presque toujours choisie, au moins pour un côté, dans les formes bilatérales.
Ils sont également de plus en plus souvent proposés dans les formes unilatérales en cas de petites tumeurs épargnant la macula laissant donc espérer une préservation visuelle de l’œil atteint, ou survenant chez un très jeune enfant, notamment dans le cadre d’un dépistage, et donc dans un contexte de risque de développer un rétinoblastome bilatéral métachrone, c’est-à-dire décalé dans le temps.
Les traitements conservateurs concernent actuellement environ 20 % des patients atteints de rétinoblastome unilatéral.
Les tumeurs en avant de l’équateur de l’œil* peuvent être traitées par cryothérapie pour les plus petites d’entre elles ou curiethérapie interstitielle (par disque d’iode ou de ruthénium radioactif), en cas de tumeur plus volumineuse ou avec un envahissement vitréen localisé.
Les tumeurs en arrière de l’équateur de l’œil peuvent être traitées par laser seul ou en combinaison avec de la chimiothérapie par carboplatine. Ce protocole permet le traitement de tumeurs du pôle postérieur sans recours à l’irradiation externe. Cette technique de thermo-chimiothérapie associe une administration de carboplatine avant réalisation d’une hyperthermie tumorale par laser diode à j1 afin d’en potentialiser les effets.
Dans le cas des atteintes très étendues de la rétine ou du vitré, le seul traitement conservateur possible reste l’irradiation externe.
Récemment, le développement de la perfusion sélective intra-artérielle de chimiothérapie est devenu une option thérapeutique notamment dans les formes localement avancées de la maladie.
* Ligne imaginaire encerclant le globe de l'œil à égale distance des pôles antérieur et postérieur.
Le suivi médical
STANDARD
La surveillance ophtalmologique se poursuit sous anesthésie générale au rythme mensuel, puis progressivement espacé, mais maintenu au moins au rythme trimestriel.
Le but de ce suivi est de dépister le plus précocement possible les récidives intraoculaires ou les nouvelles tumeurs, afin d’augmenter les chances de préservation oculaire et visuelle.
Le risque de récidive diminue avec le temps, mais des récidives ou des nouvelles tumeurs peuvent survenir même tardivement
Avec la collaboration de l’enfant, la surveillance ophtalmologique se poursuit en consultation, mais ne s’espace pas au-delà du rythme trimestriel. Cette surveillance a également pour objectifs de repérer les lésions oculaires induites par le traitement et d’évaluer précisément la vision
IMPORTANT
Tous les malades présentant un rétinoblastome doivent bénéficier d’une consultation génétique à la recherche de mutations germinales. Si elles existent, les membres de la famille se verront proposer une consultation d'oncogénétique.
LE PRONOSTIC DU RETINOBLASTOME EST BON...
Il est de nos jours excellent et de 90 à 95 % des enfants guérissent de leur maladie.
Le pronostic vital à 5 ans est identique pour la forme unilatérale et la forme bilatérale.
L’absence de récidive à 5 ans est considérée comme guérison.
En résumé
90 % des enfants sont en vie à 5 ans en l’absence de métastases
La vision est d’autant plus conservée que la tumeur est petite de taille et respecte la fovéa
Pour vous aidez
RETINOSTOP
Association de patients souffrant de rétinoblastome créée en 1994 par des parents d'enfants atteints d'un rétinoblastome. Elle est là pour vous aider et vous orienter, en particulier en ce qui concerne les démarches.
26 rue d'Ulm 75232 Paris Cedex 05 - Courriel : retinostop@retinostop.org
Mise à jour
9 mai 2015