
Les formes habituelles
Les trois formes de LLC de phénotype B
La LLC typique
Dans cette forme, la prolifération concerne des lymphocytes B matures engagés qui ont fait l'expérience de l'antigène, de la zone marginale, ressemblant à un lymphocyte B mémoire et exprimant le CD19+ et CD20+ mais ni le CD5-, ni le CD10-.
Dans ce cas, la proportion de cellules lympho-plamocytaires ou de cellules clivées est supérieure à 15 %.
La LLC atypique
Cette forme est retrouvée dans 20 % des cas. Dans ce cas il s'agit d'une prolifération de lymphocytes B matures, naïfs exprimant à la surface les protéines CD19+, CD5+, CD20+ et CD23+ ainsi que les immunoglobulines IgM et IgD.
La LLC mixte
Cette forme représente 15 % des cas de LLC. Elle est constituée d'un mélange de petits et grands lymphocytes (le cytoplasme est abondant et clair), ou essentiellement de grands lymphocytes.
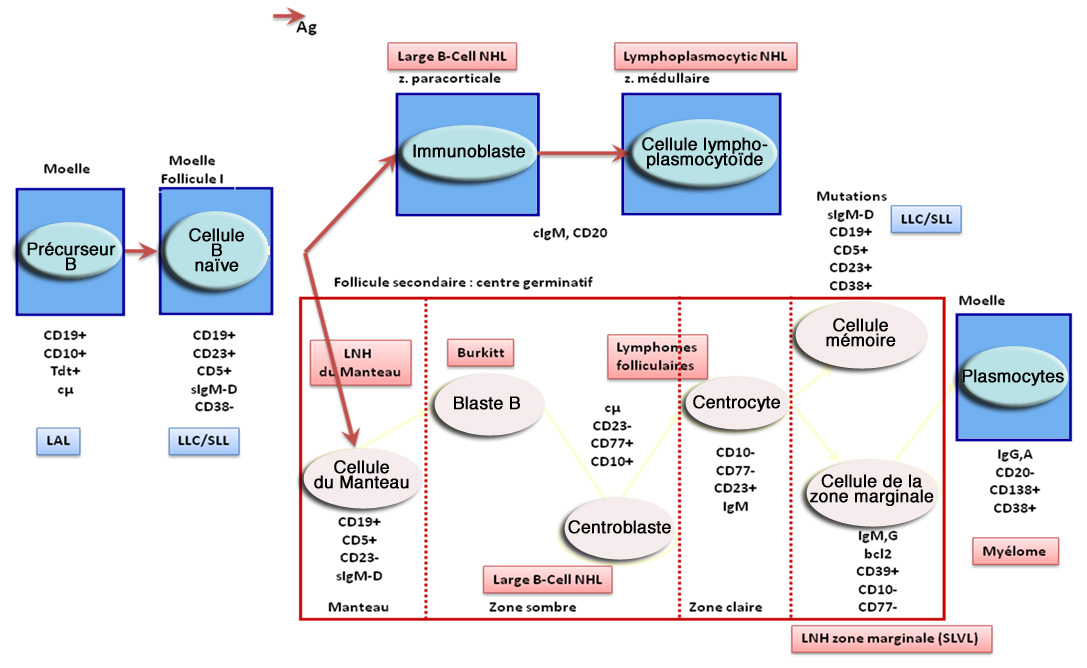
@ En cliquant sur le lien, vous retrouverez un schéma sur les formes de la maladie présentant leurs spécificités.
{kind=link}
L'importance du laboratoire...
L’hétérogénéité biologique de la LLC (état d’hypermutation des gènes des chaînes lourdes des immunoglobulines, présence d’aberrations génomiques spécifiques et/ou de mutations récurrentes dans les oncogènes et les gènes suppresseurs de tumeurs) détermine la variabilité des manifestations cliniques mais aussi et surtout l’évolution et la résistance aux différents traitement
|
Gène |
Localisation chromosomique |
Fonction |
Fréquence (%) |
Implications thérapeutiquesTraitement |
|---|---|---|---|---|
|
SF3B1 |
2q33 |
Splicéosome* |
10 à 15 % |
|
|
NOTCH1 |
9q34 |
Transduction du signal |
10 à 15 % |
|
|
BIRC3 |
11q22 |
Signalisation NFκB |
5 % |
Résistance à l’Ibrutinib |
|
ATM |
11q22 |
Dommage ADN, régulation TP53 |
10 à 20 % |
|
|
TP53 |
17p13 |
Cycle cellulaire |
7 % |
Chimiorésistance à la fludarabine |
|
RPS15 |
19p13 |
Translation |
5 % |
Chimiorésistance |
|
XPO1 |
2p15 |
Exportine |
3 % |
|
|
BTK |
Xq22 |
Signalisation BCR |
Après ibrutinib |
Résistance à l’ibrutinib |
|
PLCG2 |
16q24 |
Transduction du signal |
Après ibrutinib |
Résistance à l’ibrutinib |
|
POT |
|
Dommage ADN |
5 % |
|
(* Le splicéosome, ou particule d'épissage, est un large complexe de protéines et d'ARN qui catalyse l'élimination des introns des ARN pré-messagers et l'épissage des exons codants dans le processus de maturation des ARN messagers)
Les anomalies cytogénétiques, un aspect important de la maladie
UNE NOTION IMPORTANTE...
Comme dans toutes les formes de leucémies, la présence d’anomalies génétiques est très fréquente et on estime qu'elles sont présentent dans près de 80 % des cas. Leur recherche est maintenant systématique car l'identification du type de mutations présente un intérêt pronostique et thérapeutique. Ces mutations peuvent être mises en évidence par les techniques de cytogénétique conventionnelle.
D'autres techniques, comme l'hybridation in situ avec sonde fluorescente (FISH), la PCR multiplex de courts fragments fluorescents (QMPSF - Quantitative Multiplex PCR of Shorts Fluorescents Fragments ) ou l'hybridation génomique comparative ( CGH micro-arrays) peuvent être employées.
QUATRE TYPES PRINCIPAUX DE MUTATIONS
La délétion d'un fragment du chromosome 13q
C'est la mutation la plus fréquente qui est retrouvée dans la moitié des cas. Elle est de bon pronostic si elle demeure isolée.
La délétion d'un segment du chromosome 17p
Elle s'observe dans moins de 10 % des cas et est de pronostic très réservé. La recherche par examen cytogénétique d’une délétion 17p doit précéder toute ligne de traitement car, dans ce cas le gène p53 devient non fonctionnel. La maladie est alors résistante au traitement par la fludarabine (Fludara™).
Elle présente, elle aussi, une ré-évolutivité rapide après traitement.
La trisomie 12
Elle est plus rare et n'est retrouvée que dans environ 15 % des cas. Elle est associée à un aspect particulier des cellules.
La délétion d'un fragment du chromosome 11q
Elle est rare, 15 % des cas environ et indique un pronostic réservé.
Elle est associée à des formes tumorales de la maladie et une ré-évolutivité, rapide, après traitement.
le statut mutationnel IGHV
Récemment, la recherche de mutations somatiques au sein des régions variables VH (H pour Heavy ) du gène des immunoglobulines IgVH a conduit à l’identification de deux groupes de LLC de pronostic distinct, favorable en présence de mutations somatiques, défavorable sinon.
Il existe deux sous-types de LLC, selon le degré de mutations des gènes des immunoglobulines IgVH
- Les LLC non mutées IGHV dérivent de cellules n’ayant pas transité par le centre germinatif du ganglion lymphatique, et sont généralement plus agressives que les LLC mutées. Les cellules de LLC non mutées seraient en effet plus sensibles à l’inhibition du récepteur des lymphocyte B, BCR.
- Les LLC mutées présentent une hypermutation somatique témoignant de leur passage dans le centre germinatif du ganglion lymphatique. Elles sont à évolution plus lente et sont associé à une meilleure réponse à la chimiothérapie comportant un analogue des purines.
Mise à jour
1er juillet 2022