
Les tumeurs endocriniennes
Des maladies rares...
LEUR INCIDENCE
Les tumeurs endocriniennes ne représentent qu’un pour-cent des cancers du pancréas.
Elles surviennent, en moyenne 10 ans avant les cancers habituels du pancréas.
La plupart des tumeurs endocrines du pancréas sont, en principe, bénignes, mais il est très difficile de prédire l'évolution de ces tumeurs.
LES MALADIES CONCERNÉES
Différentes dénominations
Sous le terme "tumeur endocrinienne du pancréas" sont regroupées d'autres tumeurs comme : les tumeurs carcinoïdes, les tumeurs neuroendocrines, APUDome (APUD pour Amine Precursor Uptake and Decarboxylation).
Des tumeurs secrétant ou non...
Ces tumeurs ont comme point commun de se développer à partir de tissus embryonnaires de l’intestin antérieur. Sur un plan pratique, les spécialistes distinguent les tumeurs :
- Fonctionnelles responsables de symptômes liés à une production de substances biologiquement actives comme des peptides ou des amines. Elles nécessitent un traitement symptomatique spécifique.
- Non fonctionnelles, sans symptômes liés à une production de peptides ou d’amines.
Des tumeurs à prédisposition génétiques
Elles peuvent s’intégrer dans le spectre de maladies pour lesquelles il existe une prédisposition génétique reconnue, comme :
- Une néoplasie endocrine multiple de type 1 (NEM 1), assez souvent
- Une maladie de von Hipple-Lindau (VHL)
- Une neurofibromatose de Recklinghausen (ANR)
LEUR SIÈGE
Elles peuvent se situer sur tout le tube digestif, de l’œsophage au rectum. Les sites préférentiels sont la partie terminale de l’intestin grêle, l’iléon, l’appendice, le rectum et la région du duodénum et du pancréas.
Classification OMS 2010 des tumeurs endocrines du pancréas
Les tumeurs neuroendocrines G1
- Tumeur limitée au pancréas, sans invasion vasculaire ni infiltration péri-nerveuse : < 2cm, < 2 mitoses/10 champs à fort grossissement, ≤ 2% de cellules positives pour le Ki-67/MIB1 et un comportement évolutif bénin
- Pronostic indéterminé : tumeur limitée au pancréas, présentant une ou plus des caractéristiques suivantes : ≥ 2 cm (2-10 mitoses/10 champs à fort grossissement > 2% de cellules positives pour le Ki-67/MIB1 et invasion vasculaire et infiltration péri-nerveuse
Les tumeurs neuroendocrines G2, bien différenciées
- Bas grade de malignité mais important envahissement local et/ou métastases
- Haut grade de malignité (> 10 mitoses / 10 champs à fort grossissement)
Les autres types de tumeurs
- Les carcinomes neuroendocrines G3 à grandes ou à petites cellules peu différenciés
- Les carcinomes mixtes endocrines-exocrines (appelés adéno-neuro-endocrines)
- Les lésions hyperplasiques et pré-néoplasiques
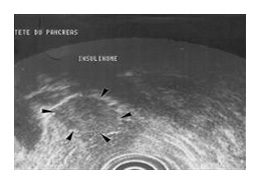
LES INSULINOMES
DES TUMEURS SECRETANTES...
Ce sont des tumeurs développées à partir cellules des îlots de Langerhans du pancréas qui contiennent des cellules spécialisées dans la production d'insuline.
Les insulinomes produisent de grandes quantités d'insuline, ce qui peut entraîner une forte baisse du taux de sucre dans le sang, c’est-à-dire des hypoglycémies surtout à jeun et à l'effort.
L'incidence est de 1 à 4 cas par million d’habitants et sont malins dans environ 10 % des cas (soit 1 à 5 nouveaux cas par an en France). Ils représentent de 35 à 45% de tumeurs endocriniennes du pancréas.
L’insulinome est souvent isolé, mais s’inscrit parfois (5 à 10 % des cas) dans le contexte d’une néoplasie endocrinienne multiple de type 1 (NEM1), où il est alors volontiers multiple.
C’est habituellement une tumeur bénigne, maligne dans 10 % des cas (la malignité ne peut être affirmée que par la survenue de métastases).
LE DIAGNOSTIC
Chez un patient ayant présenté, une glycémie <0,55g/L (spontanément ou lors d’une épreuve de jeûne) concomitante d’une insulinémie >3 mUI/L et d’un peptide C >0,6ng/mL (dite triade de Whipple) ces anomalies biologiques confirme le diagnostic d’insulinome s'il n'y a pas de prise de sulfamide et en l'absence d’anticorps anti-langerhansiens.
La tumeur est le plus souvent de petite taille (90 % font moins de 2 cm et 30 % moins de 1 cm), ce qui pose des problèmes pour la repérer en imagerie médicale. De ce fait, le diagnostic peut faire appel à une technique très particulière l’OctreoScan™.
LEUR TRAITEMENT
La chirurgie, quand elle est techniquement faisable. Souvent, d'autres options sont envisagées
- Le diazoxide (Proglicem™) ou les analogues de la somatostatine constituent les options de première ligne thérapeutique symptomatique.
- L’évérolimus est proposé en deuxième intention pour le contrôle symptomatique en cas d’intolérance ou d’échappement au diazoxide et analogues de la somatostatine.
- La chimio-embolisation hépatique d’action antisécrétoire rapide est utilisée en cas d'hypoglycémie sévère, réfractaire aux thérapeutiques médicamenteuses.
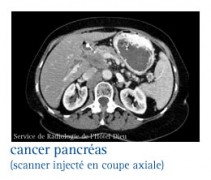
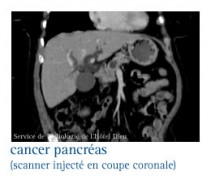
Les glucagonomes
LE SYNDROME HORMONAL
Ces tumeurs sont, en général, malignes qui produisent du glucagon en excès. Le glucagon induit une élévation très importante de la glycémie et peut induire rapidement un diabète.
Les patients présentant un glucagonome développent dans 90 % des cas une urticaire très grave appelée érythème nécrotique migratoire. Elle précède le diagnostic de 6 à 8 ans en moyenne. Elle se produit sur la partie inférieure du tronc, les fesses, le périnée et les cuisses. Les lésions évoluent sur une période de une à quatre semaines selon le cycle : macule ⇒ bulle ⇒ érosion ⇒ croûte.
Les autres manifestations comprennent : un amaigrissement (96% cas) massif, > 20kg (65% cas), des phlébite ou des manifestations thrombo-emboliques (25% cas), des diarrhées (14% cas ) non associée à un problème de malabsorption, des douleurs abdominales non spécifiques et parfois des troubles psychiques.
LE DIAGNOSTIC
Les examens biologiques révèlent une hyper-glucagonémie.
La tumeur est mise en évidence sur les échographies transpariétale et endoscopique, le scanner abdominal, voire l’artériographie cœliaque.
SON TRAITEMENT
En raison de la rareté de la pathologie, il n’existe pas de traitement bien codifié. Les options comprennent la chirurgie et la chimiothérapie.
Il repose sur la chirurgie radicale lorsqu’elle est possible
La chimiothérapie adjuvante est nécessaire lorsque la tumeur n’a pu être extirpée en totalité. Elle est traitement de base des cas inopérables et en cas de récidive ou de métastases.
Le traitement de première comporte la dacarbazine en association avec somatostatine.
En cas de métastases hépatiques localisées une embolisation artérielle hépatique est une option.
La durée du traitement est variable et dans certains cas, le traitement pourra durer deux ans.
LE GASTRINOME - SYNDROME DE ZOLLINGER-ELLISON
HISTORY
Dans la revue Ann Surg. 1955 Oct; 142(4): 709–723, deux chirurgiens américains, RM Zollinger (1903 - 1992) et EH Ellison (1918 - 1970), publient un article "Primary Peptic Ulcerations of the Jejunum Associated with Islet Cell Tumors of the Pancreas"...
UNE TUMEUR FONCTIONNELLE
Dans ces tumeurs éponymes, la gastrine est secrétée en excès. Le risque de malignité est d’environ 50 %.
L'excès de gastrine active les cellules pariétales et entérochromaffine-like de l'estomac (fundus). Ces cellules entrainent la sécrétion en excès d’acide chlorhydrique à l'origine d'ulcères gastriques ou du duodénum multiples, de diarrhées importantes et une hypertrophie de la muqueuse gastrique.
Le syndrome de Zollinger-Ellison recouvre l'ensemble des manifestations induites par une sécrétion anormale de gastrine, par une tumeur endocrine duodénale ou pancréatique (gastrinome). Il associe des ulcères duodénaux sévères et une diarrhée.
Dans certains cas, le syndrome de Zollinger-Ellison est partie intégrante d'une néoplasie endocrine multiple de type I (NEM 1), associé alors à une hyperparathyroïdie et plus rarement à d'autres atteintes des glandes endocrines.
SON TRAITEMENT
Les symptômes sont contrôlés par les médicaments antisécrétoires, en particulier les inhibiteurs de la pompe à protons, comme par exemple, le Mopral™, le Lanzor/Ogast™, l’Inexium™, l’Inipomp™, et les génériques.
La chirurgie n'est pas systématique et sa pertinence est discutée. Néanmoins, dans certains cas, la guérison peut être obtenue lorsque la tumeur et ses éventuels ganglions satellites sont accessibles à une résection radicale.
Le VIPome - Syndrome de Verner-Morrison
Le VIP
Le VIP (Vasoactive Intestinal Peptide ) est biologiquement actif au niveau des neurones intestinaux, cérébraux et de la moelle épinière, ainsi que ceux des poumons, du système urogénital et d'autres glandes endocrines.
Il est principalement responsable de la vasodilatation, de la glycogénolyse, de la lipolyse, de la résorption osseuse qui peut entraîner une hypokaliémie, de l'hypochlorhydrie, une hyperglycémie, une l'hypercalcémie, et une déshydratation.
Ces effets biologiques causés par l'augmentation de la sécrétion de VIP sont la principale cause des symptômes observés
VIPOME
Les VIPomes sont des tumeurs malignes dans 70 à 90 % des cas. Leur incidence annuelle en France est d’environ 100 nouveaux cas. Chez l'adulte, les VIPomes sont généralement diagnostiqués dans la 4ème décennie avec une légère prédominance féminine (65 % des cas)
Dans 95 % des cas les VIPomes sont isolés, 5 % de tous les cas étant associés au syndrome de néoplasie endocrinienne multiple de type 1 (NEM1). Dans plus de 60 à 80 % cas, les VIPOMES sont diagnostiqués au stade métastatique.
Le taux survie à 5 ans est de 94 % en cas de diagnostic précoce et moins de 70 % pour les formes évoluées.
LE SYNDROME HORMONAL - SYNDROME DE VERNER-MORRISON
En 1958, JV Verner (interniste) et AB Morrison (anatomopathologiste) ont décrit un syndrome associant une tumeur langerhansienne à cellules non β du pancréas, et une diarrhée hydrique profuse avec hypokaliémie. Ce syndrome Il est aussi appelé syndrome de Verner-Morrison ou encore de choléra pancréatique.
Ce syndrome est en rapport avec une tumeur maligne dans environ 60 % des cas. Elle est responsable d’une diarrhée motrice sévère (10 selles/jours), avec hypokaliémie, due à la sécrétion excessive de VIP ( Vaso-Intestinal Peptide), dosable dans le sang. Cette diarrhée persiste à l’épreuve du jeun.
Les autres manifestations sont cutanées (flush), neuropsychiques (parésie, paralysie 25%).
Les signes généraux comprennent, un amaigrissement (90% des cas) et des signes de déshydratation (80 % des cas).
SON TRAITEMENT
Le traitement repose si possible sur la chirurgie. Elle dépend du stade TNM de la maladie définit soit par l'European Neuroendocrine Tumor Society (ENETS), soit celle de l'Union for International Cancer Control (UICC).
Les analogues de la somatostatine permettent de bien contrôler la diarrhée motrice.
LE SOMATOSTATINOME
LE SYNROME HORMONAL
C'est une tumeur très rare très souvent maligne.
La tumeur se développe aux dépens cellules D soit des îlots pancréatiques, soit des cellules endocrines du tractus digestif.
Physiologiquement, la somatostatine diminue les concentrations d’insuline, de glucagon ou de gastrine.
La maladie est en association avec une neurofibromatose avec ou sans hyperparathyroïdie et hyperplasie médullaire surrénale.
Les manifestations cliniques sont inconstantes, peu spécifiques ce qui conduit souvent à des errances diagnostiques. On peut observer, un diabète, une lithiase vésiculaire, une stéatorrhée (selles blanches), un amaigrissement et une fatigue importante (asthénie).
LE DIAGNOSTIC
Le diagnostic n’est souvent fait que par un dosage systématique de la somatostatine devant une tumeur du pancréas localisée ou métastatique.
L'imagerie fonctionnelle (Octréoscan®, TEP DOTA-TOC ou DOTA-NOC) et TEP-FDG est fondamentale pour un diagnostic précis. Elle permet de réaliser le bilan d'extension de la tumeur, de donner des indications sur le pronostic — une tumeur fixant intensément sur une imagerie des récepteurs de la récepteurs de la somatostatine, sans fixation au TEP-FDG, sera de bon pronostic ; inversement, une lésion avec Octréoscan® négatif et TEP-FDG positif sera de mauvais pronostic — et peut être utile à des fins pré-thérapeutiques (radiothérapie interne vectorisée par analogues de la somatostatine radiomarqués).
LE TRAITEMENT
La stratégie thérapeutique dépend de la localisation de la tumeur primitive, du grade histopronostique, de l'évolution et du volume tumoral et la présence de métastases extra-hépatiques
Les traitements médicamenteux incluent les analogues de la somatostatine, la chimiothérapie, l’embolisation, la radiothérapie intra-artérielle hépatique, les thérapies ciblées (évérolimus) et la radiothérapie interne vectorisée.
En résumé
|
|
Gastrinome |
Insulinome |
Glucagonome |
VIPome |
Somastostatinome |
|---|---|---|---|---|---|
|
Hormone |
Gastrine |
Insuline |
Glucagon |
VIP |
Somatostatine |
|
Signes cliniques |
Ulcères duodénaux réfractaires |
Hypoglycémie survenant lors du jeune et/ou activité physique |
Erythème nécrolytique migrateur |
Diarrhées sécrétoires sévères |
Diabète |
|
Examens biologiques |
Gastrinémie à jeun élevée (>10N) |
Hypoglycémie < 0,50 g/l |
Glucagon élevé |
Vip élevé |
Somatostatine élevée |
|
Traitement |
IPP |
Chirurgie ++ |
Chirurgie |
Chirurgie |
Chirurgie |
Les tumeurs neuro-endocrines non secrétantes
Elles sont souvent malignes. Elles peuvent être bien différenciées et d’évolution lente ou indifférenciées et d’évolution rapide.
Le traitement des tumeurs bien différenciées repose sur la chirurgie si elle est possible, celui des indifférenciées sur la chimiothérapie (étoposide et cisplatine).
Les progrès récents
DE NOUVEAUX ESPOIRS...
La recherche clinique mondiale a permis de montrer l'intérêt des traitements ciblés pour ce type de pathologie.
Everolimus (Certican/Afinator™)
C'est un inhibiteur de la mTOR et actif par voie orale. Cette biothérapie, active par voie orale, donnée en traitement continu, augmente significativement la survie et le temps sans rechute de la maladie.
Il est indiqué pour le traitement dans le traitement de tumeurs neuroendocrines d’origine pancréatique non résécables ou métastatiques bien ou moyennement différenciées avec progression de la maladie.
Il est indiqué dans le traitement de tumeurs neuroendocrines d’origine gastro-intestinale ou pulmonaire non résécables ou métastatiques, bien différenciées (Grade 1 ou Grade 2), non fonctionnelles, en progression.
Sunitinib (Sutent™)
Il est homologué pour le traitement le traitement des tumeurs neuroendocrines du pancréas (pNET) non résécables ou métastatiques, bien différenciées, avec progression de la maladie chez l’adulte.
Cabozantinib (Cabometyx™)
C'est un inhibiteur de protéines kinases impliquées dans la croissance et l'angiogenèse tumorale, le remodelage osseux pathologique, la résistance aux médicaments et laprogression métastatique du cancer. Il cible le récepteur MET et des récepteurs VEGF. Il est actuellement homologué pour le traitement du cancer du rein.
Une étude de Phase-III (étude CABINET) portant sur 290 patients a montré une grande efficacité dans le traitement, en seconde ligne, des tumeurs neuroendocrines pancréatiques ou extra-pancréatiques.
UN THERANOSTIQUE
Edotreotide (SomaKit TOC™)
C'est un peptide analogue de la somatostatine, peut être radiomarqué par un nucléide émetteur β+, le gallium 68, pour effectuer une imagerie par TEP des tumeurs neuroendocrines gastro-entéro-pancréatiques bien différenciées.
177 Lu-Dotatate (Lutathéra™)
Ce peptide spécifique des cellules tumorales exprimant le récepteur à la somatostatine, a été légèrement modifié en oxodotréotide pour pouvoir être radiomarqué par le lutécium 177, émetteur β-, cette fois-ci à visée thérapeutique.
Les résultats positifs de l'étude pivot NETTER-1, portant sur 229 patients présentant une tumeur neuroendocrine digestive inopérable et/ou métastatique exprimant les récepteurs à la somatostatine, ont permet l'homologation de ce médicament.
Pour vous aider...
POUR EN SAVOIR PLUS...
Vous pouvez obtenir des informations complémentaires sur ces maladies rares en visitant les sites Internet suivants :
- En français, le site des maladies rares, ORPHANET
- En anglais pour le syndrome de Zollinger-Ellison sur le site américain du National Digestive Diseases Information et le site américain National Organization for Rare Disorders ( NORD )
POUR UNE PRISE EN CHARGE
RENATEN : Réseau National de prise en charge des Tumeurs neuro-Endocrines Malignes Rares Sporadiques et Héréditaires
DES ASSOCIATIONS
- APTED : Association des patients de tumeurs endocrines digestives Courriel : contact@apted.fr 162, avenue Lacassagne 69424 Lyon Cedex 03
- ACFE :Association Francophone de Chirurgie Endocrinienne
- CNETSC :Carcinoide NeuroEndocrine Tumor Society Canada
- GTE : Groupe d'étude des tumeurs endocrines
- WorldWide Net Cancer Day : La communauté mondiale des associations sur les tumeurs neuro-endocrines (langue anglaise)
Mise à jour
21 mars 2024