
Selon le type de tissu
Les classifications
DE L'OMS
La classification de référence des sarcomes des tissus mous est celle de l'Organisation Mondiale de la santé (OMS). Elle fut proposée initialement en 1969 et révisée en 1994.
Elle répertorie les tumeurs bénignes et malignes en 15 grands types qui sont présentés dans le tableau ci-dessous. De plus, elle décrit environ 170 sous-types de tumeurs des tissus mous.
|
LES TYPES DE TUMEURS (CLASSIFICATION OMS) |
|
|---|---|
|
Tumeurs du tissu fibreux |
Tumeurs synoviales |
D'ENZINGER-WEISS
En pratique, la classification la plus utilisée est celle d'Enzinger et Weiss qui est fondée sur l'aspect morphologique des cellules tumorales. Elle distingue 15 groupes et 58 sous-groupes de sarcomes des tissus mous.
En termes de fréquence…
Les trois types de sarcomes des tissus mous les plus fréquents sont :
- L'histiocytofibrome malin (MFH) (30%)
- Le rhabdomyosarcome, chez l’enfant et l’adolescent (15%),
- Le liposarcome (15%)
Il existe des différences de sous-types histologiques en fonction, de l'âge et du siège de la tumeur :
- Le ténosynovialosarcome chez le sujet jeune,
- Le léiomyosarcome chez le sujet âgé.

Les principaux types de tumeurs
DU TISSU FIBREUX
L'histiocytome fibrome malin
C’est la tumeur maligne des tissus mous la plus fréquente. C’est une tumeur lymphophile qui touche rapidement les aires ganglionnaires.
Au scanner, elle présente une forme polylobée, mais à bords plus ou moins bien définis avec parfois quelques calcifications et une densité hétérogène accentuée après injection du produit de contraste (iode).
Les fibrosarcomes
Les fibromes sont des tumeurs bénignes. A l’opposé, les fibrosarcomes sont des tumeurs malignes. Ils n'ont pas de caractères spécifiques en imagerie médicale.
Le diagnostic est fait par l’examen des tissus provenant de la biopsie, au microscope.
La fibromatose musculo-aponévrotique ou tumeur desmoïde
Elle se voit surtout chez l'adulte jeune au niveau des grosses masses musculaires, la fesse en particulier.
Son caractère est infiltrant et elle a tendance à récidiver.
Elle est associée à un comportement très variable. Il existe une mutation du gène APC et de la béta caténine. De ce fait, le bilan impliquera la recherche de polypose familiale
DU TISSU GRAISSEUX
Les lipomes
Ce sont les tumeurs bénignes très fréquentes. Elles peuvent être uniques, multiples ou rentrant dans le cadre d'une lipomatose.
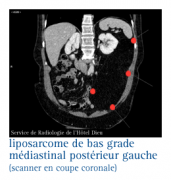
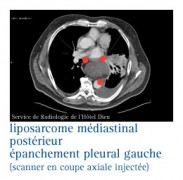
Les liposarcomes
Ce sont des tumeurs malignes beaucoup plus rares que les tumeurs bénignes. Ils se rencontrent entre 25 et 55 ans.
Dans plus de 60 % des cas, ils siègent au niveau du membre inférieur ; plus rarement, ils peuvent être rétropéritonéaux.
Les spécialistes distinguent 4 variétés histopathologiques de liposarcomes :
- Les formes bien différenciées
- Les formes à cellules rondes
- La forme myxoïde (5 % des cas) avec un pic d'incidence à 50 ans
- La forme plésiomorphe
Le pronostic est fonction du grade histologique (G1‐G2 : bon pronostic, peu de risque de métastases ; G3‐G4, risque de métastases pulmonaires.
DES MUSCLES
Il existe deux variétés de tumeurs musculaires, en fonction du tissu d’où émerge la tumeur.
Du tissu musculaire strié
Ce sont essentiellement les muscles des membres qui sont affectés.
Les rhabdomyomes sont des tumeurs bénignes rares et siègent avec prédilection dans la région cervico-faciale et chez l'enfant. Les rhabdomyosarcomes sont des tumeurs malignes.
Du tissu musculaire lisse
Les léiomyomes sont des tumeurs bénignes superficielles. Les léiomyosarcomes sont des tumeurs malignes. Cette tumeur est chimiosensibles et il y a rarement de métastases à distance.
DE LA SYNOVIALE
La synovite villo-nodulaire pigmentée ou hémopigmentée
C’est une maladie très rare. Elle possède de nombreux synonymes : synovite nodulaire, ténosynovite nodulaire, bursite villo-nodulaire pigmentée, xanthome des gaines.
En pratique, les spécialistes distinguent 2 formes.
La forme diffuse se développe au dépends de la synoviale articulaire des genoux, de la hanche, du tarse.
La forme nodulaire est parfois articulaire, mais elle se développe, surtout, au niveau de la synoviale des gaines des tendons des extrémités (tumeur à cellules géantes des gaines- xanthome -ténosynovite) ou des bourses séreuses.
La synovite villo nodulaire pigmentée du genou, à la différence des autres localisations, se comporte comme une tumeur des parties molles.
Le synovialosarcome
Il se voit entre 15 et 30 ans et affecte préférentiellement le genou. Il se présente dans la très grande majorité des cas comme une tumeur extra articulaire tant sur le plan clinique que radiologique.
Il reste classé dans les tumeurs synoviales alors qu'il semble de plus en plus qu'il soit issu d'une cellule mésenchymateuse multipotente avec une translocation spécifique aboutissant, dans les deux tiers des cas, à un gène de fusion SYT/SSX1 .
Son pronostic est réservé en raison de la fréquence des récidives locales.
Les tumeurs à cellules géantes des gaines tendineuses et les tumeurs téno-synoviales à cellules géantes
Ce sont des proliférations tumorales touchant la synoviale des cavités articulaires et des tendons.
DU CARTILAGE ET DES TISSUS MOUS
Les chondromes des tissus mous
Ils sont rares. Ils sont parfois suggérés sur les radiographies par l'existence de calcification, de densité variable, en volutes ou en anneaux.
Les chondromes extra osseux peuvent coexister avec un chondrome ou une chondromatose osseuse.
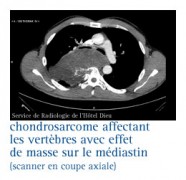
Les chondrosarcomes
Ils sont encore plus rares. La distinction d'avec le chondrome est difficile.
Les ostéosarcomes primitifs des tissus mous
Ils sont extrêmement rares et tout comme d'ailleurs les chondrosarcomes, posent un problème de diagnostic et donc de conduite à tenir, souvent difficile, avec la Myosite Ossifiante Circonscrite (MOC). La MOC, affection bénigne, comprend 2 variétés.
- Post-traumatique
- Pseudo-tumorale pour laquelle l'on ne retrouve pas de traumatisme et qui inclut les para-ostéo arthropathies neurogènes (POA).
La MOC, affection bénigne, plus fréquente chez le sujet jeune, évolue en 3 stades :
- Aigu de type inflammatoire (4 à 5 semaines)
- Chronique (4-6 mois)
- Régression avec disparition des symptômes (1 à 2 ans)
LES MÉTASTASES
Les métastases dans les tissus mous de tumeurs primitives sont rares. Les mélanomes et les cancers bronchiques seraient les tumeurs primitives les plus fréquemment en cause.
@ Pour vous y retrouver dans la dénomination compliquée des types de tumeurs cliquez ici, sur : DÉNOMINATION DES TUMEURS
Pour les tissus spécialisés...
DU TISSU NERVEUX
Le neurinome
C'est une tumeur bénigne nerfs périphériques appelée, aussi, schwannome. Il est, en général, solitaire, comme le neurinome du nerf acoustique. Les schwannomes malins se rencontrent entre 30 et 60 ans.
Les neurofibromes
Ils souvent multiples, comme dans la neurofibromatose de type 1 (maladie de Recklinghausen) et la neurofibromatose de type 2 (centrale). On observe une transformation maligne dans environ 10 % des cas.
Les neurofibrosarcomes
Ce sont des tumeurs malignes très rares
VASCULAIRES
Les angiosarcomes
Ce sont des tumeurs rares d’évolution agressive représentant environ 1 à 2% des sarcomes des tissus mous. Elles sont des tumeurs de haut grade. Leurs sites de prédilection incluent la peau, en particulier de la tête et le scalp, le sein, la rate et les os.
Les facteurs de risque reconnus sont un antécédent de radiothérapie et/ou de lymphœdème.
Le développement de la maladie peut être favorisée par un œdème chronique congénital ou acquis, comme un lymphœdème après la chirurgie d'un cancer du sein (angiosarcome de Stewart et Treves), des sites d’irradiation, une ulcération chronique….
En dépit, des progrès dans la connaissance de la maladie, le pronostic reste sombre avec une survie globale à cinq ans de l'ordre de 40 %. Les traitements locaux, lorsqu’ils sont possibles, comprennent une chirurgie complétée par une radiothérapie adjuvante. En phase métastatique, les traitements de première ligne incluent la doxorubicine ou le paclitaxel hebdomadaire.
Les hémangiomes
Ils sont classés selon la taille et l'épaisseur des parois des vaisseaux qui les constituent. On décrit des hémangiomes capillaires, caverneux, mixtes et veineux.
Les hémangioendothéliomes
Ils doivent leur nom à la prolifération des cellules à l’intérieur des vaisseaux, appelées cellules endothéliales.
Les hémangiopéricytomes
Ce sont des tumeurs qui se développent à partir des péricytes. Ces formes bénignes ont leurs correspondances sarcomateuses dont la fréquence est bien moindre.
Les lympho-angiosarcomes et les lymphangiomes
Les vaisseaux lymphatiques sont à l'origine de ces tumeurs.
Les classifications biomoléculaires
LES SARCOMES AVEC UN PROFIL GÉNÉTIQUE DÉFINI...
Les sarcomes avec translocation spécifique
Dans ce cas, cette anomalie peut être mise en évidence par différentes techniques et permettent un diagnostic précis de la maladie et, souvent, se traduisent par la mise en œuvre d'un traitement ciblé.
Les sarcomes avec un profil génomique simple caractérisé par la présence d’amplifications
Cela concerne essentiellement les liposarcomes, bien différenciés ou dédifférenciés, et les sarcomes intimaux (intima - partie interne des vaisseaux).
Ces tumeurs présentent une amplification des gènes MDM2 et CDK4 associée, parfois, à une amplification d’autres gènes.
Les sarcomes avec une mutation activatrice
Dans environ 90 % des tumeurs stromales gastro-intestinales (GIST) , il existe une mutation d’un gène codant pour un récepteur à tyrosine kinase (RTK) , KIT et/ou PDGFRA.
La mise en évidence de ces mutations est utile pour prédire la réponse à l’imatinib et également pour expliquer la résistance secondaire à l’imatinib.
Les sarcomes avec mutation inactivatrice
Cela concerne certaines tumeurs du muscle strié (tumeur rhabdoïde maligne) chez laquelle on détecte une inactivation complète du gène INI1 .
LES AUTRES SARCOMES
Ce sont, encore, les plus courants...
Ils présentent un profil génomique complexe caractérisé par de nombreux gains et des pertes géniques, avec fréquemment perte du gène RB1 et altération de p53.
Lesquels ?
Les léiomyosarcomes, rhabdomyosarcomes pléomorphes, liposarcomes pléomorphes, myxofibrosarcomes et sarcomes peu différenciés (histiocytofibromes malins et fibrosarcomes) appartiennent à cette catégorie et ne montrent pas d’anomalie spécifique.
Mise à jour
28 septembre 2022