
Les syndromes myélodysplasiques (SMD)
DES ÉTATS PRÉ-LEUCÉMIQUES
DE QUOI S'AGIT-IL ?
Ce sont des maladies de la moelle osseuse
Elles sont caractérisées par une hématopoïèse inefficace aboutissant à une diminutions des éléments du sang périphérique (cytopénie). Les deux caractéristiques principales de cette maladie sont une incidence qui augmente avec l'âge et une évolution vers une leucémie aiguë myéloïde dans environ 30 % des cas.
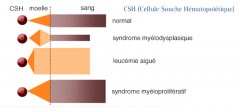
C'est une hémopathie monoclonale
Elle affecte une cellule souche hématopoïétique multipotente commune aux trois lignées caractérisée, à de degrés divers, par :
- Une destruction importante anormale des globules rouges (hémolyse) intramédullaire
- Une hématopoïèse inefficace qui ne peut plus produire suffisamment une, deux ou les trois sortes de cellules normales du sang périphérique
- Des cytopénies progressives, débouchant sur une anémie arégénérative, une neutropénie ou une thrombopénie
- Une moelle osseuse généralement riche à la ponction biopsie osseuse (BPO)
- La production de cellules anormales, dites myélodysplasiques, portant sur une ou plusieurs lignées
- Un taux de blastes (cellules immatures) variable
Plus récemment, des anomalies du gène TET2 porté par le chromosome 4 ont été identifiées et sont communes à l'ensemble des maladies de ce syndrome.
LES CHIFFRES...
Le taux d’incidence pour 100 000 personnes‑années monde est de 3,4 chez l’homme et de 1,6 chez la femme, soit un rapport hommes/femmes égal à 2,1.
En France, le nombre de cas en 2018 estimé est de 4 750 dont 60 % chez l’homme.
Ce sont des hémopathies malignes du sujet âgé, avec un âge médian au diagnostic de 78 à 80 ans. Les deux tiers des patients ont plus de 75 ans.
QUELLES EN SONT LEURS CAUSES ?
LES FACTEURS DE RISQUE
La cause précise des syndromes myélodysplasiques reste inconnue, à ce jour
Constitutionnels
Il existe des prédispositions génétiques dans un tiers des cas de l’enfant et plus rarement chez l’adulte jeune.
Les principales causes chez l’enfant sont des maladies génétiques comme la trisomie 21, l’anémie de Fanconi, la neurofibromatose de type I et d'autres syndromes très rares.
Rarement, des mutations germinales des gènes RUNX1, des gènes de la télomérase TERC et TERT, du gène GATA2 et du gène DDX41 entraînent également un risque élevé de développer la maladie à l’âge adulte.
Environnementaux
Fumer augmente le risque de LAM (d’un facteur de 1,6 environ) et peut-être le risque de SMD.
La consommation quotidienne de boissons alcoolisées peut faire diminuer le nombre de globules rouges et de plaquettes mais n’est pas à l’origine des SMD.
La radiothérapie et la chimiothérapie (agents alkylants et inhibiteurs de la topoisomérases II) peuvent jouer un rôle déclencheur.
L’incidence des syndromes myélodysplasiques (SMD) est augmentée chez les patients porteurs de maladies auto-immunes/inflammatoires. En parallèle, certains traitements immunosuppresseurs sont associés à la survenue de SMD, principalement l’azathioprine.
SECONDAIRE OU PRIMAIRE...
On fait une distinction entre les SMD primaires (sans cause connue) et les SMD secondaires (associés à un agent leucémogène). Dans 15 à 20 % des cas les SMD sont secondaires à :
- Une agression de l'ADN ou à sa fragilité : chimiothérapies avec agents alkylants (melphalan, cyclophosphamide, …) et les inhibiteurs des topoisomérases II (étoposide, doxorubicine, mitoxantrone, ....)
- Une exposition professionnelle au benzène, aux solvants organiques, aux dérivés du pétrole, aux radiations ionisantes
Dans ces cas, on retrouve souvent des anomalies chromosomiques dans la moelle osseuse et la maladie évolue plus souvent vers une LAM.
Les signes et le diagnostic
RIEN
Environ la moitié des patients ne présentent aucuns signes. Ils sont dits asymptomatiques. La maladie est découverte à l'occasion d'une prise de sang.
UNE ANÉMIE
Les SMD se présentent fréquemment par un manque de globules rouges (anémie), d'où sa dénomination ancienne d'anémie réfractaire. L’anémie est qualifiée de réfractaire en raison de fait qu’elle n’est pas améliorée par un traitement à base de fer.
L’anémie est aussi arégénérative puisque les cellules souches des globules rouges ne se développent pas proportionnellement pour corriger l’anémie. C’est un trouble rare de l’hématopoïèse appelé dysmyélopoïèse qui se caractérise par des anomalies qualitatives et quantitatives des lignées myéloïdes.
D'AUTRES SIGNES
On peut observer, en dehors de l'anémie :
- Des signes généraux
- Une anémie associée à une thrombopénie ou une leucopénie
- Une grosse rate ou splénomégalie
- Des problèmes dermatologiques
- Une pathologie dysimmunitaire : des vascularites, une polychondrite atrophiante , des dermatoses neutrophiliques
LE DIAGNOSTIC
La constatation de cytopénies, de la présence de formes anormales ou d'une monocytose supérieure à 1000 sur l'hémogramme (numération-formule sanguine NFS) doit conduire votre médecin à vous proposer la réalisation d'un myélogramme.
Le myélogramme révèle une moelle riche, avec cellules hématopoïétiques dysplasiques. Ceci permet de faire la différence avec les cytopénies à moelle pauvre (aplasie médullaire) ou à moelle envahie (par les blastes d'une leucémie aiguë ou une métastase cancéreuse) et les anémies par carence vitaminiques (B9 ou B12) qui présentent une moelle riche mais porteuse d'anomalies spécifiques (mégaloblastes).
La classification de la FAB
L’anémie réfractaire sans sidéroblaste (AR)
C’est une anémie dite réfractaire car elle n'est pas corrigée par un traitement à base de fer (traitement martial). Elle se caractérise la présence de moins de 15 % de sidéroblastes dans la moelle osseuse. Cette anémie, dans 15 % des cas, se transforme en leucémie aiguë.
L’anémie réfractaire avec sidéroblastes (ARS)
Cette forme est caractérisée par plus de 15 % de sidéroblastes dans la moelle osseuse. Elle a la même propension à se transformer en leucémie aiguë.
L’anémie réfractaire avec excès de blastes (AREB)
Elle se caractérise par l’existence d’une anémie et la présence de plus de 5 % et de moins de 20 % de blastes dans la moelle osseuse. Au bout de 5 à 10 ans, dans un tiers des cas elle se transforme en leucémie aiguë.
L’anémie réfractaire avec excès de blastes, en transformation (AREB-t)
Elle est définie par l’existence d’une anémie et la présence de 20 à 30 % de blastes au myélogramme. Ceci est en relation avec un blocage de maturation des progéniteurs conduisant à un excès chronique de cellules blastiques dans la moelle. Elle se transforme, assez rapidement, en leucémie aiguë.
La leucémie myélomonocytaire chronique
C’est une forme frontière entre les syndromes myéloprolifératifs et les myélodysplasies.
Cette maladie se caractérise par une augmentation du taux de globules blancs (hyperleucocytose) portant sur la lignée monocytaire associée à une grosse rate (splénomégalie). Le myélogramme montre une augmentation du taux des monocytes médullaires. Dans environ 30 % des cas, cette leucémie chronique se transforme en leucémie aiguë.
La classification de l'OMS (2008)
LE PRINCIPE
Elle prend en compte 5 paramètres et insiste sur le fait que le nombre de lignées atteintes est plus important que la cytopénie spécifique :
- Le nombre de cytopénies périphériques : plaquettes < 100 000, polynucléaires neutrophiles < 1 800 et une hémoglobine < 10 g/L
- Le pourcentage de blastes circulants et médullaires
- La présence de sidéroblastes en couronne (> 15 %)
- La présence éventuelle de corps d’Auer
- Une anomalie cytogénétique, la délétion 5q isolée
LES DIFFÉRENTES MALADIES
- Anémie réfractaire (AR) ; avec sidéroblastes en couronne (ARS)
- Cytopénie réfractaire avec dysplasie multilignée (CRDM)
- Cytopénie réfractaire avec dysplasie multilignée et sidéroblastes en couronne (CRDM - RS)
- Anémie réfractaire avec excès de blastes (AREB) : AREB-1 (blastes médullaires: 5 - 9%) ou AREB-2 (blastes médullaires: 10 - 19%)
- Syndromes myélodysplasiques non classables
- Syndrome 5q
Les facteurs pronostiques
Le score pronostique IPSS (International Prognostic Scoring System - Blood 1997;89:2079-88)
Il prend en compte les données suivantes:
- L'importance de la ou des cytopénies :
- Le taux d'hémoglobine: < 10 g définissant l'importance de l'anémie
- La neutropénie < 1 500 polynucléaires neutrophiles
- La thrombopénie < 100 000 plaquettes
- Les anomalies génétiques retrouvées sur le caryotype :
- Favorable : normal ou Y, del(5q), del(20q)
- Défavorable : anomalies du chromosome 7, complexes avec 3 ou plus anomalies
- Intermédiaire : autres anomalies
- Dans la moelle osseuse, le taux de blastes (formes immatures des éléments figurés du sang) : <5 % versus de 5 à 10 % et versus de 11 à 19 %
Le niveau de risque
Il est ainsi défini par un score
- Faible : 0
- Intermédiaire - 1 : 0.5 - 1.0
- Intermédiaire - 2 : 1.5 - 2.0
- Élevé : > 2.5
IPSS-R 2012
| Groupes pronostic | Anomalie | Patients concernés |
|---|---|---|
| Très bon | Chromosome Y , del(11q) | 5 % |
| Bon | Caryotype normal, del(5q), del(12p),del(20q) ou 2 anomalies dont la del(5q) |
65 % |
| Intermédiaire | del(7q), +8, +19, i(17q) Toute autre anomalie simple ou double |
15 % |
| Médiocre | -7, inv(3)/t(3q)/del(3q) Deux anomalies dont -7/del(7q) Caryotype complexe avec 3 anomalies |
5% |
| Sévère | Caryotype complexe avec > 3 anomalies | 10 % |
L'évolution de la maladie
Les SMD n’abrègent pas forcément l’espérance de vie car la moelle osseuse défectueuse suit une évolution très progressive.
Néanmoins, dans certains cas, la moelle osseuse est dans l’incapacité totale de produire des cellules normales. Les patients ne peuvent alors plus combattre les infections, du fait du manque de globules blancs, ni arrêter les saignements, du fait du manque de plaquettes ; une anémie survient également.
Dans un tiers des cas environ elle peut évoluer vers une la leucémie aiguë myéloïde (LAM).
Les objectifs des traitements proposés
Vous appartenez au groupe de risque faible ou intermédiaire : objectif du traitement : améliorer la qualité de vie
(corriger les manques de globules blancs et/ou rouges et/ou plaquettes (cytopénies) et surtout l’anémie)
Vous faites partie du groupe de risque élevé : objectif du traitement : retarder l’évolution de la maladie.
LES TRAITEMENTS Symptomatiques
LES TRANSFUSIONS DE SANG
Les indications
Les patients du groupe à faible risque ou à risque modéré ne produisent pas assez de globules rouges. Dans ce cas, vous recevrez des transfusions pour maintenir un taux d’hémoglobine normal. Si le taux d’hémoglobine (Hb) est inférieur à 8 g/L, on vous transfusera deux unités par jour à répéter à intervalle régulier. Ces transfusions sont répétées chaque fois que votre taux d’hémoglobine descend en dessous de 8 g/L.
Les inconvénients
Les globules rouges apportent du fer. Après 10 à 20 transfusions, ce fer se dépose dans le cœur et dans le foie, risquant d’entraîner des complications. Il peut alors être nécessaire d’administrer un chélateur du fer qui est un médicament pour faire baisser le taux de fer dans le sang comme :
- La déféroxamine (Desféral™) active par voie sous-cutanée ou intraveineuse
- La défériprone (Ferriprox™) et le déférasirox (Exjade™) actifs par voie orale
LES AUTRES INTERVENTIONS
Erythropoïétine ou autres agents stimulant l’érythropoïèse (ASE)
L’EPO, produite par les reins, aide au développement de globules rouges. Son but est de prévenir l’anémie pour éviter les transfusions sanguines. Toutefois, elle n’est active que dans 15 % des cas environ. Son effet est plus marqué lorsque le taux d’EPO sanguin de base est peu élevé.
En France, elle peut être prescrite à travers un protocole temporaire de traitement (PTT). De fortes doses sont nécessaires de 30 à 80 000 UI par semaine par voie sous-cutanée d’époïétine alpha ou bêta, ou 150 à 300 μg par semaine de darbepoétine. Le délai de réponse est de quatre à huit semaines, la valeur cible d’hémoglobine se situe entre 11 et 12 g/L.
Reblozyl™ (luspatercept)
C'est un inhibiteur du TGF-β actif par voie injectable.
De fait, une des causes de l’inhibition de l’érythropoïèse est l’augmentation de la super famille des TGF-β.
Initialement évalué dans la thalassémie avec des résultats très intéressants, il a, par la suite, été étudié chez des patients SMD-LR dépendant des transfusions en échec d’EPO, avec des résultats positifs.
En France, il dispose d'une autorisation temporaire d'utilisation (ATU) dans cette indication.
GSF
Le G-CSF est une protéine qui contribue à la production de globules blancs par la moelle osseuse.
Le G-CSF (Neupogen™, Granocyte™, biosimilaires), administré par voie sous-cutanée plusieurs fois par semaine, augmente le nombre de globules blancs et, de ce fait, pourrait réduire les risques d’infection ultérieure. Le G-CSF serait surtout utile chez les patients ayant un taux bas de globules blancs et qui souffrent d’infections répétées. Néanmoins, i l faut savoir que les indications de ce ce traitement ne font pas consensus.
FACE À UNE DIMINUTION DU TAUX DE PLAQUETTES
Actuellement, il n’existe pas de médication très active sur la thrombopénie de ces maladies.
Les transfusions de plaquettes sont rarement pratiquées, sauf dans les cas où le nombre de plaquettes est inférieur à 10 000 et en cas d'hémorragie. En général, les patients développent une résistance à ce traitement au bout de quelques mois.
L'autre option est un traitement par les hormones mâles (androgènes) qui permettent, parfois, d’améliorer le taux de plaquettes.
Plus récemment des traitements, utilisant des facteurs de croissance agissant sur les plaquettes ont été mis au point. Il s'agit d'agonistes du récepteur à la thrombopoïétine (TPO).
- Le romiplostime (Nplate™) actif par voie injectable (IV ou s.c.)
- L'eltrompag (Promacta™), actif par voie orale
A ce jour, ces deux médicaments ne sont pas encore homologués pour cette indication (seulement dans le traitement du purpura thrombopénique).
U?E GREFFE DE MOELLE
La greffe de moelle osseuse ou de cellules souches sanguines guérit les patients en détruisant toutes leurs cellules myélodysplasiques. Cependant, c’est un traitement lourd, qui peut entraîner des complications, et qu’il est difficile d’effectuer au-delà de 55 ans, sauf dans des conditions très particulières. Les SMD touchant les personnes âgées en priorité, la greffe n’est donc pas réalisable dans la majorité des cas.
Les traitements spécifiques
LES MÉDICAMENTS HYPOMÉTHYLANTS
L’azacitidine (AzaC, 5-AZA, AZA) est un inhibiteur de la méthylation de l’ADN, classé parmi les antimétabolites, a ctif par voie injectable sous cutanée.
Ce médicament est homologué, en France, pour le traitement des patients adultes non éligibles pour une transplantation de cellules souches hématopoïétiques et présentant un syndrome myélodysplasique de risque intermédiaire 2 ou élevé selon l'index pronostique international ( International Prognostic Scoring System , IPSS).
La dose de 75 mg/m² est administrée en cycles de sept jours consécutifs, tous les 28 jours. Ce traitement nécessite une prophylaxie pour éviter les vomissements par un médicament anti-HT 3 (sétron) avec ou sans Emend TM.
Les effets secondaires, en dehors d'irritation au site d'injection, sont hématologiques et digestifs.
C’est une molécule proche l’azacitidine qui est homologuée, aux USA pour les mêmes indications thérapeutiques que le Vidaza TM
En Europe, et en France, cette molécule n'est homologuée que pour le traitement de la leucémie myéloïde aiguë..
Le médicament est actif par voie injectable intraveineuse. L‘administration se fait sous forme d’une perfusion de 3 heures de 15 mg/m², trois jours consécutifs, toutes les 6 semaines.
Ce médicament immuno-modulateur, proche du thalidomide est actif par voie orale.
Il est indiqué pour le traitement des patients présentant une anémie avec dépendance transfusionnelle due à un syndrome myélodysplasique à risque faible ou intermédiaire associé à une anomalie cytogénétique de type délétion 5q isolée, lorsque les autres options thérapeutiques sont insuffisantes ou inappropriées.
Imatinib (Glivec™ & génériques)
Glivec ™ est indiqué dans le traitement des patients adultes atteints de syndromes myélodysplasiques/myéloprolifératifs (SMD/SMP) associés à des réarrangements du gène du PDGFR (platelet-derived growth factor receptor) .
Demain
L'imelstat, oligonucléotide, inhibiteur enzymatique de l'activité de la télomérase est prometteur.
UNE CHIMIOTHÉRAPIE DE LEUCÉMIE
Chez les patients classés à haut risque et à risque intermédiaire, la maladie peut se transformer en leucémie aiguë. Dans ce cas, on vous proposera une chimiothérapie, le plus souvent par voie injectable et en hospitalisation du type de celle utilisée pour le traitement des leucémies aiguës.
Les traitements selon le stade de la maladie
SMD DE FAIBLE RISQUE (IPSS < 1,5)
Traitement de l'anémie
Dans le cas général les transfusions représentent un traitement symptomatique. Le taux d’hémoglobine en-deçà duquel il est recommandé est 10 g/dl ou en cas d’anémie mal tolérée, quel que soit le chiffre d’hémoglobine. L’objectif est de maintenir un taux cible d’hémoglobine ≥ 10 g/dl.
Les agents stimulants l’érythropoïèse (EPO) sont utilisés chez les patients qui ont des taux d’EPO bas et un faible besoin transfusionnel. Ils sont proposés aux patients qui ont un taux d’EPO < 200 UI/ml et qui reçoivent moins de 2 transfusions par mois.
Le traitement est débuté en utilisant des posologies de 40000 à 60000 unités pour l’érythropoïétine alpha et de 300 μg/semaine pour la darbepoïétine alpha.
Les alternatives thérapeutiques sont :
- Les androgènes, comme le danazol ou la noréthandrolone, permettent d’obtenir des réponses.
- Les immunosuppresseurs sont essentiellement réservés aux patients de moins de 60 ans avec un SMD à moelle pauvre, faiblement transfusés et ayant un groupage HLA de type HLA DR-15.
Le traitement de la thrombopénie
Elle est présente chez environ 10 % des patients au diagnostic. La thrombopénie est le plus souvent d’origine centrale mais une origine périphérique ou mixte est possible offrant alors des choix thérapeutiques plus simples comme la corticothérapie ou les immunoglobulines polyvalentes. Les autres options sont :
- Les transfusions de plaquettes de concentré plaquettaire d’aphérèse
- Les androgènes
- Les agonistes du récepteur de la thrombopoïétine (TPO), eltrombopag et romiplostim.
Le traitement de la neutropénie
Il n’existe à l’heure actuelle aucun traitement standard recommandé de la neutropénie. La prise en charge repose sur le traitement précoce et adapté des complications infectieuses. Certaines équipes préconise l'utilisation de facteurs de croissance médullaire.
LES SMD DE RISQUE ÉLEVÉ (IPSS ≥ 1,5)
L’objectif principal des traitements
Il est de prolonger la survie en allongeant le délai entre le traitement reçu et la progression de la maladie.
Les thérapeutiques actuelles ont permis d’atteindre cet objectif de manière statistiquement significative. De plus, quelque soit le traitement spécifique mis en place, les traitements symptomatiques et les transfusions gardent une place majeure.
Les agents hypométhylants
Vidaza™
Les indications sont les SMD à risque élevé définis par un score IPSS ≥ 1,5, les leucémies aigües avec un pourcentage de blastes compris entre 20 et 30 % et avec dysplasies multilignées ainsi que les leucémies myélo-monocytaires chroniques (LMMC) avec un pourcentage de blastes compris entre 10 et 29 %, sans évidence pour un syndrome myéloprolifératif.
La posologie usuelle est de 75 mg/m²/j pendant 7 jours consécutifs.
Dacogen™
C' est le deuxième agent hypométhylant disponible pour le traitement des SMD de risque élevé. Le schéma d’utilisation est de 20 mg/m²/8h par voie IV sur 3 heures, pendant 3 jours avec des cycles se répétant toutes les 6 semaines.
Chimiothérapie intensive
C'est alors le même traitement que celui d'une leucémie aigüe myéloblastique (LAM). L’association habituelle est celle d’une anthracycline sur 3 jours associée à de l’aracytine sur 7 jours.
Allogreffe
C'est le seul traitement curatif dans les SMD de risque élevé chez les sujets jeunes.
L’allogreffe myélo-ablative a une efficacité démontrée dans les SMD. L’allogreffe, après conditionnement non myélo-ablatif (mini-greffe), est moins toxique que l’allogreffe classique, mais elle est associée à un risque de rechute plus élevé.
Pour les patients de moins de 50 ans en bon état général bénéficient peuvent être candidats à une allogreffe classique. Les sujets âgés de plus de 50 ans ou porteurs de comorbidités peuvent bénéficier d’une allogreffe à conditionnement atténué.
LES SYNDROMES 5Q- (DÉLÉTION 5Q ISOLÉE [DEL 5Q])
Ils se voient plus volontiers chez la femme. Ils associent fréquemment une anémie profonde, macrocytaire, un taux de PNN souvent normal et un taux de plaquettes normal voire volontiers élevé. L’évolution vers une LAM est rare et la survie souvent prolongée (médiane de 116 mois).
Le traitement des syndromes 5q- est essentiellement celui de l’anémie, voire des autres cytopénies lorsqu’elles existent. En l’absence de cytopénie ou de retentissement clinique, il n’y pas d’indication pour traiter.
Mise à jour
14 novembre 2023